
近日,《Journal of Advanced Research》(中科院一区Top期刊,影响因子:13)在线发表了题为“Snapin Mediates Neuronal PANoptosis after Mild Traumatic Brain Injury via H2S-Dependent S-sulfhydration of CTSD”的研究论文。该研究以苏州大学基础医学院为第一完成单位,陈学士、黄鑫琪、安钰梅为第一作者,张明阳教授为最后通讯作者。本研究首次揭示了Snapin蛋白在轻度创伤性颅脑损伤(mTBI)后通过调节硫化氢(H₂S)代谢稳态,进而诱导神经元发生PANoptosis的关键机制,为mTBI后的神经保护提供了全新的治疗靶点和理论依据。

创伤性颅脑损伤(TBI)是导致神经功能障碍与长期残疾的重要原因,其中mTBI因临床表现隐匿、病理机制复杂,且长期缺乏有效的干预靶点,一直是研究的难点与重点。传统研究多聚焦于凋亡、焦亡等单一形式的程序性细胞死亡,而忽视了不同死亡通路间的协同作用。近年来提出的PANoptosis概念,强调了凋亡、焦亡与坏死性凋亡可在同一细胞内整合激活,然而其在mTBI神经元损伤中的具体作用及上游调控机制尚不清楚。
张明阳教授团队长期致力于脑外伤后继发性损害的机制研究。该研究系统揭示mTBI后Snapin蛋白在皮层与海马神经元中显著上调,并伴随典型PANoptosis分子标志物的同步激活。通过构建神经元特异性敲低Snapin的腺相关病毒(AAV-shSnapin)进行干预,证实抑制Snapin能够显著减轻脑外伤后神经退行性病变、减少PANoptosis相关蛋白表达,并有效促进小鼠在运动、认知及学习记忆等多方面的神经功能恢复。
机制研究发现,mTBI后Snapin可与内源性H₂S合成的关键酶——胱硫醚β-合酶(CBS)相互作用,扰乱脑内H₂S代谢稳态,导致具有神经保护作用的内源性H₂S水平下降。H₂S的减少削弱了其对溶酶体天冬氨酸蛋白酶CTSD前体(pro-CTSD)的S-巯基化修饰。这种翻译后修饰的减弱,促进了pro-CTSD向活性成熟形式(m-CTSD)的转化。活化的m-CTSD最终触发了神经元内PANoptosis的发生。研究团队还通过药理学手段验证了该通路的可干预性:给予外源性H₂S供体(NaHS)或CTSD抑制剂(pepstatin A)治疗,均可显著抑制CTSD成熟、减轻PANoptosis与神经炎症,并改善长期神经功能预后。
本研究系统地揭示了Snapin-H₂S-CTSD轴在调控mTBI后神经元PANoptosis中的核心作用(示意图见下图),阐明了Snapin在mTBI后由“代偿性调节因子”向“致病性调控因子”转变的分子机制。这项工作不仅深化了对脑外伤后复杂细胞死亡网络的理解,更为脑外伤后针对Snapin、恢复H₂S稳态或抑制CTSD活性等治疗策略提供了重要的理论依据,具有显著的临床转化前景。
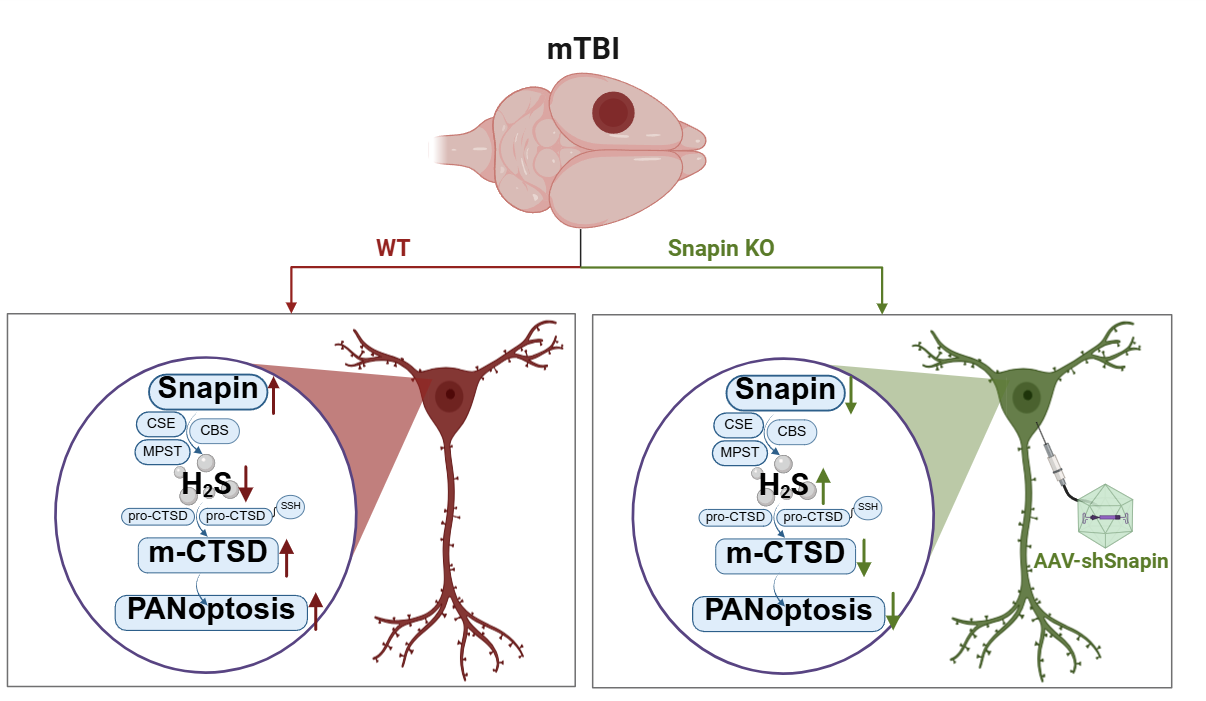
图:Snapin介导mTBI后神经元PANoptosis的作用机制示意图
本研究得到了国家自然科学基金面上项目、可园高层次人才项目等基金的资助。
原文链接:
https://www.sciencedirect.com/science/article/pii/S2090123226000640